Chapter 4 Bioinformatics resources
In this chapter some of the most important Bioinformatics resources are described. An attempt to organized them by topic is made, but most of these resources are highly integrated. So, for example, you would thing that to do a Blast search you need to go to the Blast web page. But you can also perform Blast searches from the Uniprot and EnsEMBL web pages, to mention just two. Similarly, most if not all sequence databases link the associated Gene Ontology information to their sequence entries. This high degree of integration might make difficult at first to value the existence of different resources.
4.1 Sequence databases
NCBI
The National Center for Biotechnology Information (NCBI) host many sequence databases including genome databases and the Genbank repository for sequence information. It also host many Bioinformatics resources like the Blast tool.
Figure 4.1: Screenshot of the NCBI home page at https://www.ncbi.nlm.nih.gov.
EnsEMBL
EnsEMBL is a database where the genome information about many species is collected with a focus con comparative genomics research.
Figure 4.2: Screenshot of the EnsEMBL home page at https://www.ensembl.org.
Uniprot
The Uniprot website focuses on information about protein sequences, including splice forms.
Figure 4.3: Screenshot of the Uniprot home page at http://www.uniprot.org.
4.2 Gene expression databases
GEO: Gene Expression Omnibus
The Gene Expression Omnibus (GEO) database collects data from high-throughtput experiments including microarrays and NGS datasets.
Figure 4.4: Screenshot of the GEO home page at https://www.ncbi.nlm.nih.gov/geo/.
The number of submissions per year for microarrays and RNA-seq continues to increase.
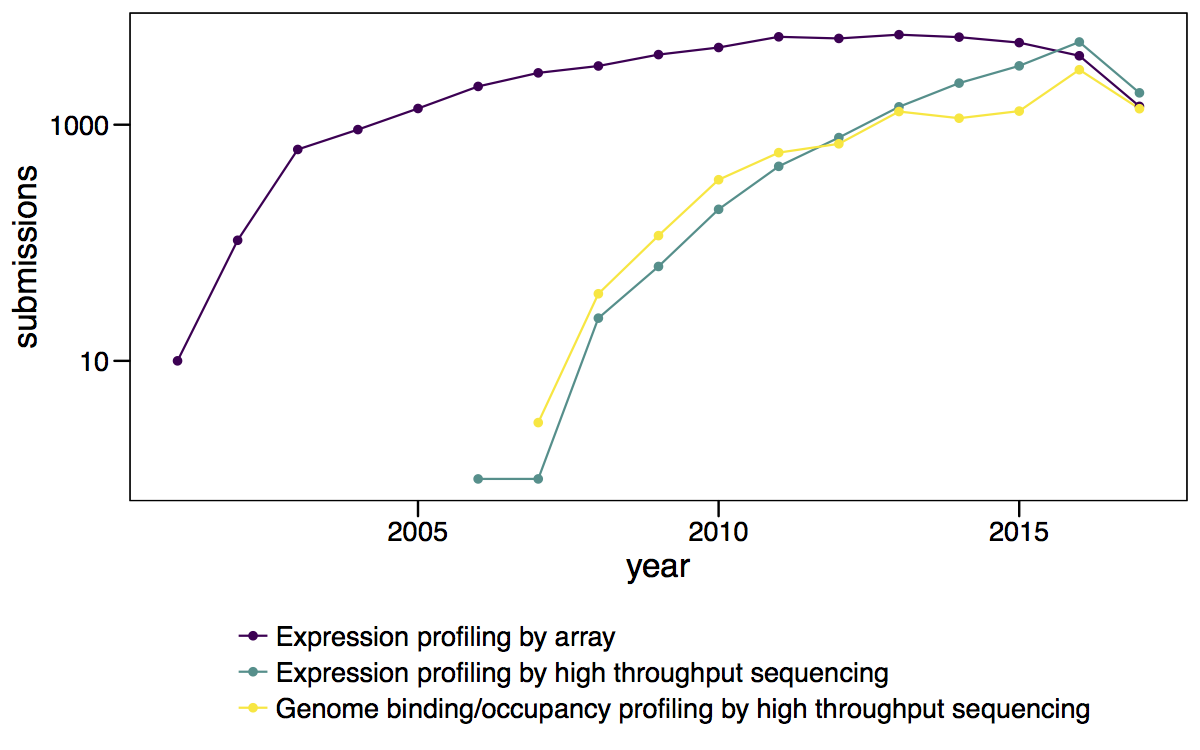
Figure 4.5: Number of GEO submissions per year for the top 4 technologies.
ArrayExpress
ArrayExpress is the alternative at the European Bioinformatics Institute.
Figure 4.6: Screenshot of the ArrayExpress home page at https://www.ebi.ac.uk/arrayexpress/
4.3 Pathway databases
As information about genes, proteins and metabolites accumulates, it becomes more and more important to organize this knowledge in a comprehensive way. We have mentioned how genes and their products participate in the networks of cellular processes, the cell’s pathway. This network includes metabolic, signaling and gene regulatory networks. This information is critical to understand the cell’s phenotype, but the accumulated knowledge is daunting. It would benefit to have some central repository with easily accessible tools that enables to visualize existing knowledge in an easy way. It would be also very useful if it was possible to use that information in computer analysis. This is the motivation behind some of the web resources described below.
KEGG
KEGG stands for Kyoto Encyclopedia for Genes and Genomes (Figure 4.7). Is an integrative resource with information about matabolic and signaling pathways in different species, from bacteria and archaea to mammals.
Figure 4.7: Screenshot of the KEGG home page at http://www.kegg.jp.
Reactome
4.4 Domain databases
Pfam
Pfam is a database of protein domain families.
Proteins are generally comprised of one or more functional regions, commonly termed domains. The presence of different domains in varying combinations in different proteins gives rise to the diverse repertoire of proteins found in nature. Identifying the domains present in a protein can provide insights into the function of that protein.
The Pfam database is a large collection of protein domain families. Each family is represented by multiple sequence alignments and a hidden Markov model (HMMs).
Figure 4.8: Screenshot of the Pfam home page at http://pfam.xfam.org.
4.5 Motif databases
Jaspar
Jaspar is a database for transcription factor DNA binding motifs.
Figure 4.9: Screenshot of the Jaspar home page at http://jaspar.genereg.net.
4.6 Interaction databases
Information about protein-protein interactions has grown incredibly due to advances in MS in combination with affinity purification methods. There are several databases integrating existing information. Two resources will be highlighted here: BioGRID and STRING.
BioGRID
BioGRID focuses on storing information about PPIs for which experimental evidence exists.
Figure 4.10: Screenshot of the BioGRID home page at https://thebiogrid.org.
STRING
STRING combines experimental and computational evidence to infer PPIs.
Figure 4.11: Screenshot of the STRING home page at https://string-db.org.
4.7 Other
Gene Ontology
One important difficulty when dealing with Biological structures (molecules, organelles, etc.) is the vocabulary we use to described them. As this type of information accumulated different researches would tend to use slighly different ways to describe the same thing. In other cases they can use similar words to describe different things. These nuances make the descriptions often found in publications ambiguous making it difficult to determine the exact meaning of some assertion. This difficulty became more prominent once the amount of information increased dramatically with the advent of omics technologies, and the information about the biological stuff was the subject of computational analyses. There was a need for an standardized nomenclature, and that was the origin of the Gene Ontology project. The purpose of the Gene Ontology project is stated in it web page:
The Gene Ontology (GO) project is a major bioinformatics initiative to develop a computational representation of our evolving knowledge of how genes encode biological functions at the molecular, cellular and tissue system levels. Biological systems are so complex that we need to rely on computers to represent this knowledge. The project has developed formal ontologies that represent over 40,000 biological concepts, and are constantly being revised to reflect new discoveries. To date, these concepts have been used to “annotate” gene functions based on experiments reported in over 100,000 peer-reviewed scientific papers.
In particular:
The Gene Ontology project provides a controlled vocabulary of terms for describing gene product characteristics and gene product annotation data from GO Consortium members, as well as tools to access and process these data.
Figure 4.12: Screenshot of the Gene Ontology home page at http://www.geneontology.org.
4.8 Bioformatics software
4.8.1 Sequence analysis
Blast
The Blast web page at NCBI is the main point of access to this versitile tool. It can be used to search for sequence similarity against any of the sequence databases at the NCBI.
Figure 4.13: Screenshot of the Blast home page at https://blast.ncbi.nlm.nih.gov.
HMMER
HMMER implements Hidden Markov Models for the analysis of biological sequences. It can learn models representing motifs, domains or entire sequences. These models are used to search for sequences matching the models in databases. It can also be used to annotate the domains/motifs present in a sequence using a database of domains. PFAM uses HMMER and manually curated MSA of domains to generate a database of protein families (PFAM). The use of HMM for sequence alignment is described in detail in the excellent book Biological sequence analysis: Probabilistic models of proteins and nucleic acids.
Figure 4.14: Screenshot of the Hmmer home page at https://www.ebi.ac.uk/Tools/hmmer.
Jalview
Jalview is a software for sequence analysis. It can visualize sequences, submit them to multiple sequence aligment servers, visualize the resulting MSA, create basic phylogenetic trees and visualize associated structures.
Figure 4.15: Screenshot of the Jalview home page at http://www.jalview.org.
MEME
The MEME-suite is a collection of tools for the discovery of motifs in proteins, DNA and RNA sequences. If can find de-novo motifs (e.g. MEME) or search for known motifs from a motif database (e.g. MAST, FIMO, etc.)
Figure 4.16: Screenshot of the MEME-suite home page at http://meme-suite.org.
The meme commdan line tool enables to search for de novo motifs in nucleotide or protein sequences. The basic usage is:
meme -o <output_folder_name> -nmotifs <n> <fasta file>Where <output_folder_name> is the name of the output directory where we want our results to be placed, <n> is the maximum number of motifs we want meme to search for (default if -nmotifs ommited is 1), and <fasta file> is the file in FASTA format with our sequences.
fimo enables to scan a database of sequences in a FASTA file for motifs from a motif database. The motif database could be the output from meme (for example, the meme.txt file inside the output directory). It can also be a database from other motif databases, like Jaspar. These need to be formatted appropriately before using it with fimo. The MEME suite includes a jaspar2meme command line tool that enables to format the Jaspar database in a way suitable to use with fimo.
fimo <motif file> <fasta file>4.8.2 Next generation sequencing
Bowtie2
Bowtie2 is a command line software to align sequence reads obtained from NGS technologies to a reference genome. It requires sequencing data in Fastq format and produces aligments in SAM format.
Figure 4.17: Screenshot of the Bowtie2 home page at http://bowtie-bio.sourceforge.net/bowtie2/index.shtml.
Samtools
The samtools is a command line software for the manipulation of SAM files. It can convert SAM files into binary (BAM) files and perform other tasks like sorting and indexing.
Figure 4.18: Screenshot of the samtools home page at http://www.htslib.org.
4.8.3 General frameworks
Bioconductor
Bioconductor is a project providing R packages for the anlysis of omics datasets in the R software. R runs on Windows, Macs and Linux computers.
Bioconductor provides tools for the analysis and comprehension of high-throughput genomic data. Bioconductor uses the R statistical programming language, and is open source and open development.
Figure 4.19: Screenshot of the Bioconductor home page at http://www.bioconductor.org.
Cytoscape
Cytoscape is a software for the analysis and visualization of networks. It enables to import network information from online databases and integrate it with omics data like for example gene expression profiles. Cytoscape runs on Windows, Macs and Linux computers.
Figure 4.20: Screenshot of Cytoscape showing the HIV sample dataset http://www.cytoscape.org.
Galaxy
The Galaxy project provides a web platform for the analysis of NGS data. In their own words:
Galaxy software framework is an open-source application (distributed under the permissive Academic Free License. Its goal is to develop and maintain a system that enables researchers without informatics expertise to perform computational analyses through the web. A user interacts with Galaxy through the web by uploading and analyzing the data. Galaxy interacts with underlying computational infrastructure (servers that run the analyses and disks that store the data) without exposing it to the user.
Figure 4.21: Screenshot of the main Galaxy site at http://usegalaxy.org.
4.9 File formats
A summary of several file formats useful in genomic research is given here.
FASTA
FASTA is a very simple file format used to store information about biological sequences, including protein, DNA and RNA. Each entry consists of the > character followed by a sequence Id and a description, separated from the Id by a space character. The sequence itself goes in the next lines. Often FASTA files are formatted with a maximum number of residues per line (e.g. 60), which helps when visualizing the information in a regular text editor.
>seq1 This is seq1
AGTTAGGATTTGCGCCATTFastq
FASTQ is a format used to store information from NGS technologies. In particular, stores the sequences of the reads in addition to information about the quality of sequencing. Each entry starts with the @ character. That line also contains other information like the read identifier. The next line is the sequence information. Next there is a line with the + character, and then is the quality information encoded as a phred score of the same length as the sequence.
@K00211:114:H52N5BBXX:1:2125:22252:26301
TTCCTATATAATCTTTATTTCTGGAAGTTAAAGTAGTTACAGCAATATACAAAAACAAACAACAACAAAAAACAACCCACAATAATATAAATTTTTACAC
+
AAFFFJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJFJJJJJJJJJJJJJJJJJJJJJJSAM
Sequence Aligment/Map format is a text format to store the alignment of NGS reads to the reference genome. The basic fields are described in the help file, for which a screenshot is shown below. The full specification can be found here.
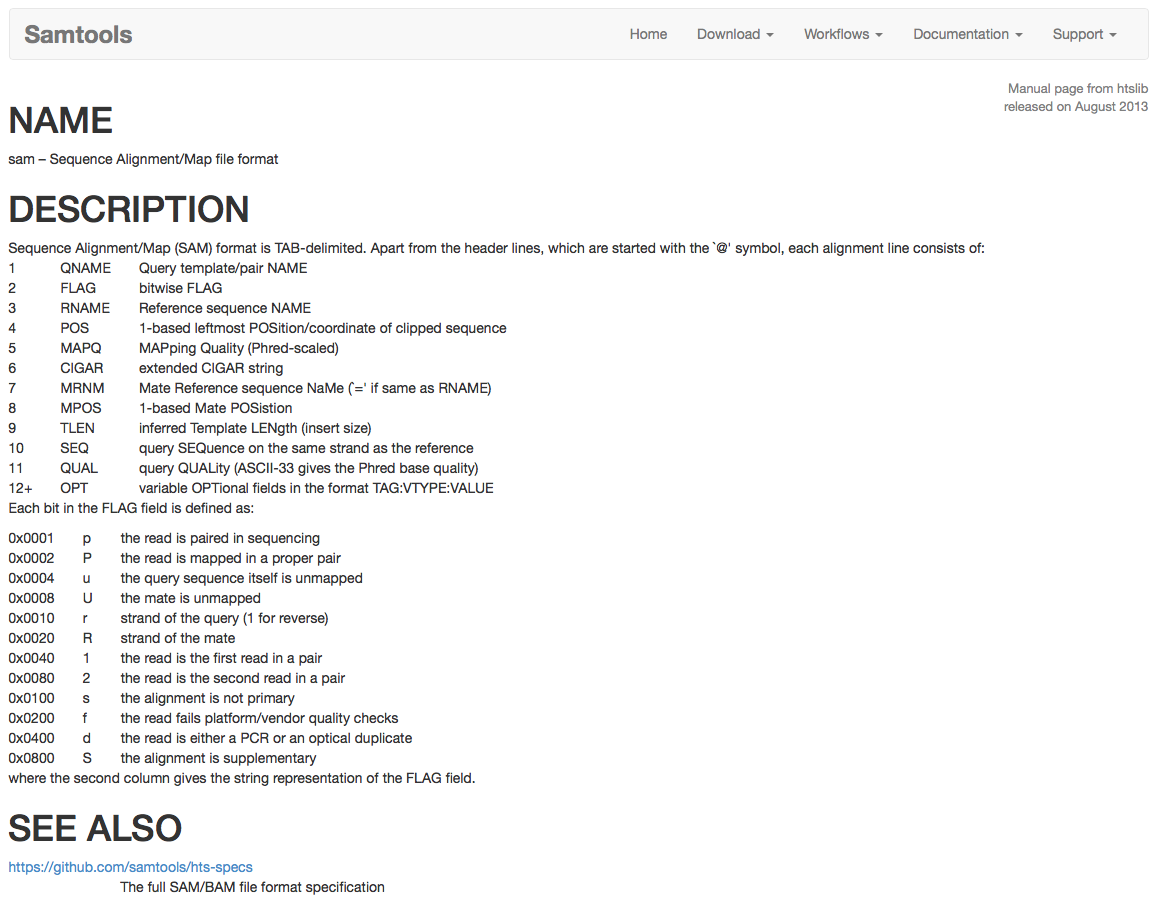
Figure 4.22: Screenshot of the SAM file format basic specification found at http://www.htslib.org/doc/sam.html.
BED
Browser Extensible Data (BED) format is used to represent genomic features like exons, transcripts, genes, etc. Each line contains at least the following columns: chromosome, start, end, name, score and strand.
chr7 127471196 127472363 Pos1 0 +
chr7 127472363 127473530 Pos2 0 +
chr7 127473530 127474697 Pos3 0 +
chr7 127474697 127475864 Pos4 0 +
chr7 127475864 127477031 Neg1 0 -
chr7 127477031 127478198 Neg2 0 -GFF
General Feature Format (GFF) is used to represent genomic features like exons, transcript, genes, etc.
browser position chr22:10000000-10025000
browser hide all
track name=regulatory description="TeleGene(tm) Regulatory Regions"
visibility=2
chr22 TeleGene enhancer 10000000 10001000 500 + . touch1
chr22 TeleGene promoter 10010000 10010100 900 + . touch1
chr22 TeleGene promoter 10020000 10025000 800 - . touch2